---
title: "5. Bayesian Hierarchical Joint Modeling for Historical Data Borrowing"
author:
- Daniel Sabanés Bové
- Francois Mercier
date: last-modified
editor_options:
chunk_output_type: inline
format:
html:
code-fold: show
html-math-method: mathjax
cache: false
---
The purpose of this document is to show a minimal workflow for Bayesian hierarchical joint modeling for historical data borrowing using the `jmpost` package.
## Setup and load data
Here we execute the R code from the setup and data preparation chapter, see the [full code here](0_setup.qmd).
```{r}
#| label: setup_and_load_data
#| echo: false
#| output: false
library(here)
options(knitr.duplicate.label = "allow")
knitr::purl(
here("session-bhm/_setup.qmd"),
output = here("session-bhm/_setup.R")
)
source(here("session-bhm/_setup.R"))
knitr::purl(
here("session-bhm/_load_data.qmd"),
output = here("session-bhm/_load_data.R")
)
source(here("session-bhm/_load_data.R"))
```
## Descriptive analysis of the current study data
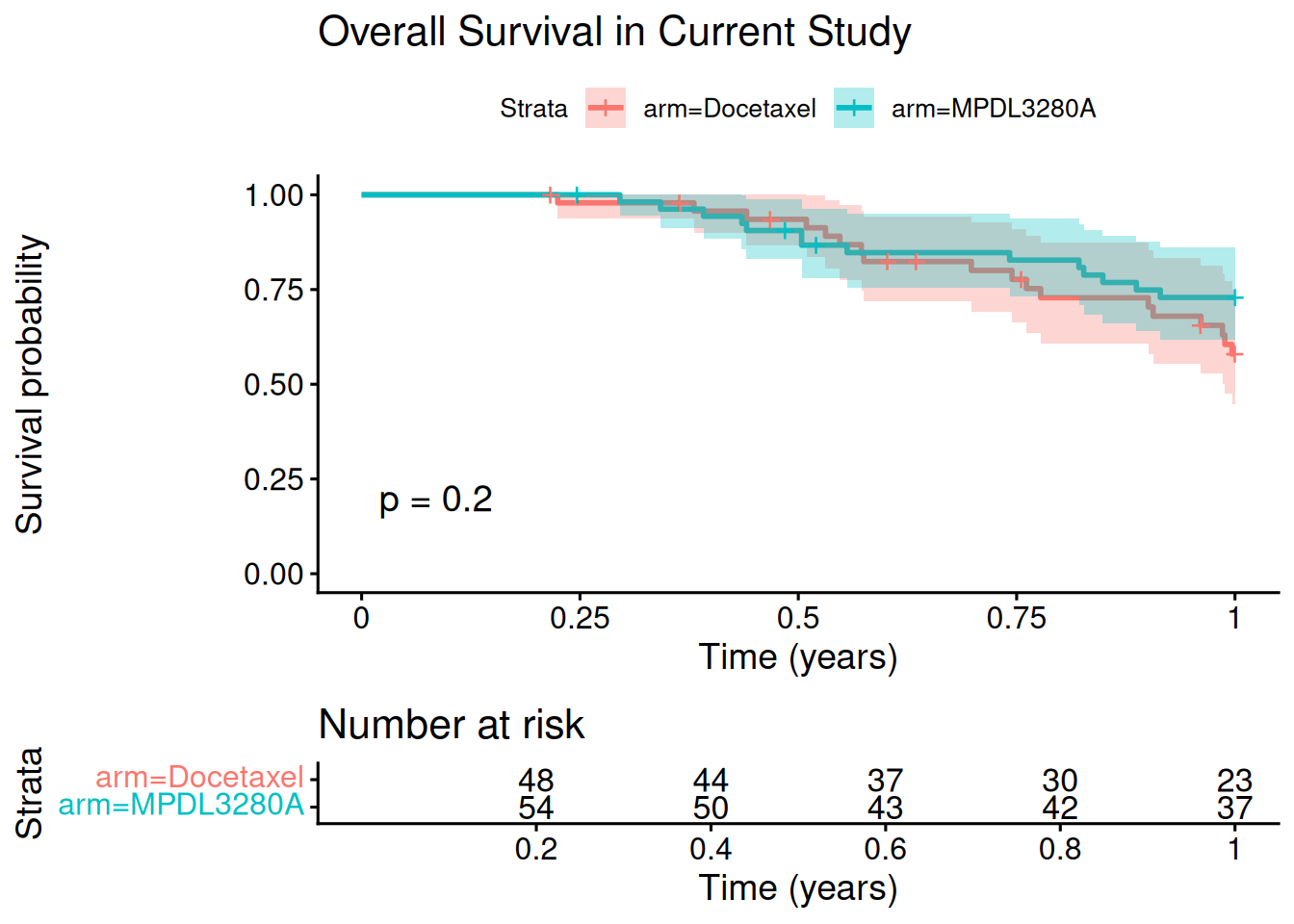
Let's have a quick look at the (artificially reduced to maximum 1 year follow-up) current study data, in order to illustrate the typical situation where we have immature OS data:
```{r}
#| label: descriptive_current_data
library(survival)
library(survminer)
# Overall Survival KM plot
fit_os_current <- survfit(Surv(os_time, os_event) ~ arm, data = current_os_data)
ggsurvplot(
fit_os_current,
data = current_os_data,
risk.table = TRUE,
pval = TRUE,
conf.int = TRUE,
xlab = "Time (years)",
title = "Overall Survival in Current Study"
)
```
So no matter which model we might fit to this data, we will not be able to trust it to estimate the hazard ratio reliably.
## Check compatibility of historical and current data
Just to mention that we would need to double check the compatibility of historical and current clinical trial design (including patient populations, endpoints, assessment schedules, etc.) and data distributions before applying any kind of historical data borrowing technique.
In our dummy example here, this is fulfilled by construction.
## Fitting the joint model only to the historical data
The first step is to fit the joint model only to the historical data. In reality, this also includes model selection in order to determine:
- choice of parametric baseline survival model (log-logistic, Weibull, etc.?)
- selection of baseline covariates
- choice of the TGI metric to correlate with the survival model, i.e. the link function (SLD, tumor growth rate, time to tumor regrowth, etc.?)
We have discussed model comparison and selection in session 3 [here](https://rconis.github.io/tgi-os-training/session-jm/1_jm-jmpost.html#model-comparison), so we will skip this step here and directly fit a joint model to the historical data only.
```{r}
#| label: fit_joint_model_historical
historical_subj_df <- historical_os_data |>
select(study, id, arm)
historical_subj_data <- DataSubject(
data = historical_subj_df,
subject = "id",
arm = "arm",
study = "study"
)
historical_long_df <- historical_tumor_data |>
select(id, year, sld)
historical_long_data <- DataLongitudinal(
data = historical_long_df,
formula = sld ~ year
)
historical_surv_data <- DataSurvival(
data = historical_os_data,
formula = Surv(os_time, os_event) ~ ecog + age + race + sex
)
historical_joint_data <- DataJoint(
subject = historical_subj_data,
longitudinal = historical_long_data,
survival = historical_surv_data
)
historical_joint_mod <- JointModel(
longitudinal = LongitudinalSteinFojo(
mu_bsld = prior_normal(log(65), 1),
mu_ks = prior_normal(log(0.52), 1),
mu_kg = prior_normal(log(1.04), 1),
omega_bsld = prior_normal(0, 3) |> set_limits(0, Inf),
omega_ks = prior_normal(0, 3) |> set_limits(0, Inf),
omega_kg = prior_normal(0, 3) |> set_limits(0, Inf),
sigma = prior_normal(0, 3) |> set_limits(0, Inf)
),
survival = SurvivalWeibullPH(
lambda = prior_gamma(0.7, 1),
gamma = prior_gamma(1.5, 1),
beta = prior_normal(0, 20)
),
link = linkGrowth(
prior = prior_normal(0, 20)
)
)
options("jmpost.prior_shrinkage" = 0.999)
save_file <- here("session-bhm/histmod1.rds")
if (file.exists(save_file)) {
historical_joint_results <- readRDS(save_file)
} else {
historical_joint_results <- sampleStanModel(
historical_joint_mod,
data = historical_joint_data,
iter_sampling = ITER,
iter_warmup = WARMUP,
chains = CHAINS,
parallel_chains = CHAINS,
thin = CHAINS,
seed = BAYES.SEED,
refresh = REFRESH
)
saveObject(historical_joint_results, file = save_file)
}
```
Let's have a look at the results:
```{r}
#| label: summarize_historical_model
vars <- c(
"lm_sf_mu_bsld",
"lm_sf_mu_ks",
"lm_sf_mu_kg",
"lm_sf_sigma",
"lm_sf_omega_bsld",
"lm_sf_omega_ks",
"lm_sf_omega_kg",
"beta_os_cov",
"sm_weibull_ph_gamma",
"sm_weibull_ph_lambda",
"link_growth"
)
mcmc_historical_joint_results <- cmdstanr::as.CmdStanMCMC(historical_joint_results)
mcmc_historical_summary <- mcmc_historical_joint_results$summary(vars)
print(mcmc_historical_summary, n = 30)
```
Let's also look at the covariate effect estimates again:
```{r}
os_cov_name_mapping <- function(surv_data) {
surv_data_design <- as_stan_list(surv_data)$os_cov_design
os_cov_names <- colnames(surv_data_design)
old_coef_names <- as.character(glue::glue("beta_os_cov[{seq_along(os_cov_names)}]"))
setNames(old_coef_names, os_cov_names)
}
os_cov_renaming <- os_cov_name_mapping(historical_surv_data)
draws_historical_joint_results <- mcmc_historical_joint_results$draws(vars)
draws_historical_joint_results <- do.call(
rename_variables,
c(list(draws_historical_joint_results), os_cov_renaming)
)
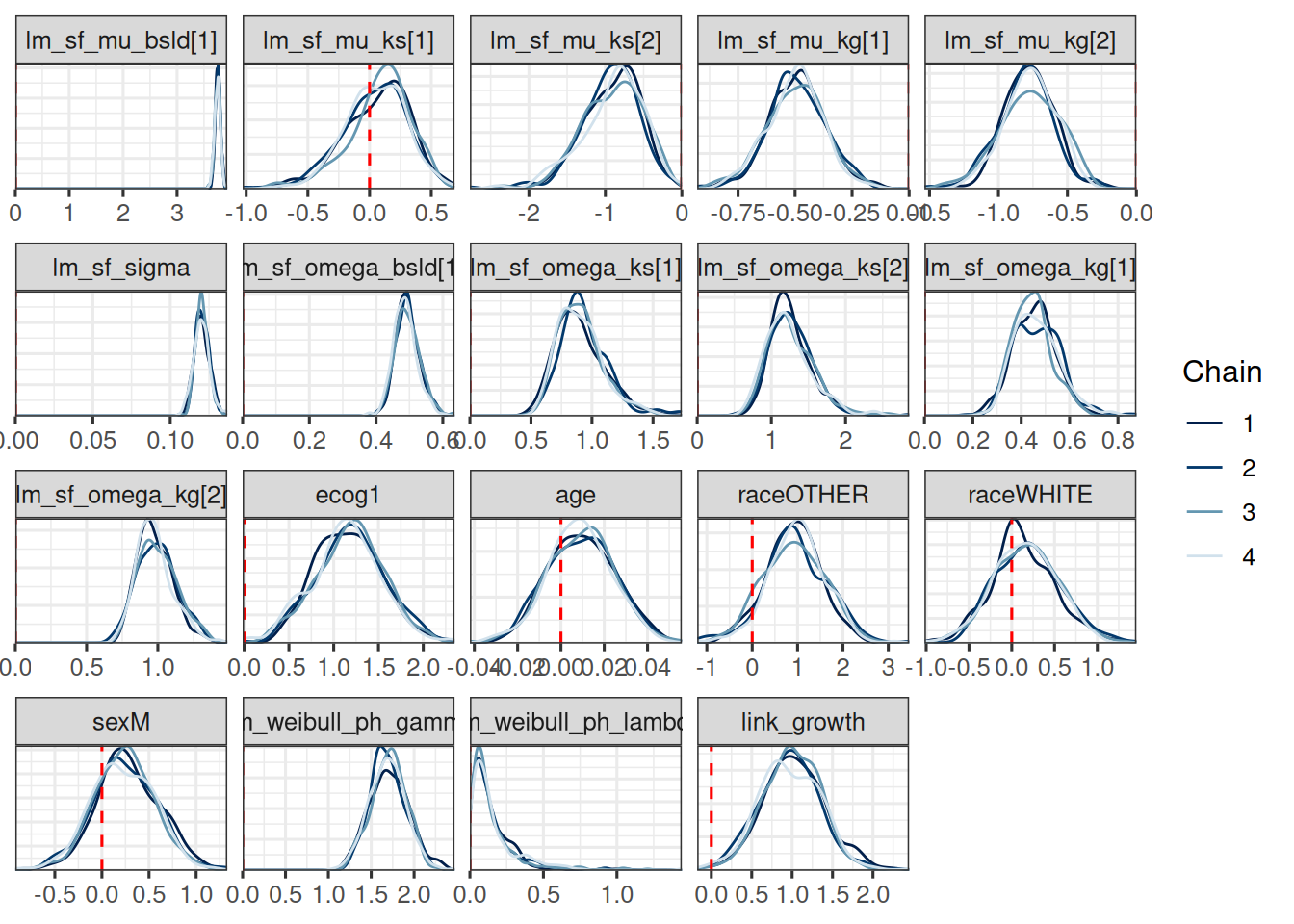
mcmc_dens_overlay(draws_historical_joint_results) +
geom_vline(xintercept = 0, linetype = "dashed", color = "red")
```
## Pool historical and current data
Now we pool the historical and the current data for the hierarchical joint modeling step:
```{r}
#| label: pool_data
pooled_os_data <- bind_rows(
historical_os_data,
current_os_data
)
pooled_tumor_data <- bind_rows(
historical_tumor_data,
current_tumor_data
)
pooled_subj_df <- pooled_os_data |>
select(study, id, arm)
pooled_subj_data <- DataSubject(
data = pooled_subj_df,
subject = "id",
arm = "arm",
study = "study"
)
pooled_long_df <- pooled_tumor_data |>
select(id, year, sld)
pooled_long_data <- DataLongitudinal(
data = pooled_long_df,
formula = sld ~ year
)
pooled_surv_data <- DataSurvival(
data = pooled_os_data,
formula = Surv(os_time, os_event) ~ ecog + age + race + sex
)
pooled_joint_data <- DataJoint(
subject = pooled_subj_data,
longitudinal = pooled_long_data,
survival = pooled_surv_data
)
```
So now we have pooled our `r nrow(historical_os_data)` historical and `r nrow(current_os_data)` current patients into a single data set with `r nrow(pooled_os_data)` patients.
## Fit the Bayesian hierarchical joint model
Now we can fit the Bayesian hierarchical joint model to the pooled data set, borrowing information from the historical data to inform the current data analysis.
Note that we use the same model specification `historical_joint_mod` as for the historical data only model fit above.
The reason is that the `LongitudinalSteinFojo` class already accounts for hierarchical modeling, see the statistical specification [here](https://genentech.github.io/jmpost/main/articles/statistical-specification.html#stein-fojo-model).
```{r}
#| label: fit_pooled_joint_model
save_file <- here("session-bhm/pooledmod1.rds")
if (file.exists(save_file)) {
pooled_joint_results <- readRDS(save_file)
} else {
pooled_joint_results <- sampleStanModel(
historical_joint_mod,
data = pooled_joint_data,
iter_sampling = ITER,
iter_warmup = WARMUP,
chains = CHAINS,
parallel_chains = CHAINS,
thin = CHAINS,
seed = BAYES.SEED,
refresh = REFRESH
)
saveObject(pooled_joint_results, file = save_file)
}
```
Let's first check again if the MCMC sampling went fine:
```{r}
#| label: check_pooled_model
mcmc_pooled_joint_results <- cmdstanr::as.CmdStanMCMC(pooled_joint_results)
mcmc_pooled_summary <- mcmc_pooled_joint_results$summary(vars)
print(mcmc_pooled_summary, n = 30)
```
Indeed all `rhat` values are close to 1 indicating convergence.
## Investigating the parameter estimates
We can see that now we have more model parameters, let's see which are additional compared to the historical data only model:
```{r}
#| label: compare_pooled_historical
pooled_param_names <- mcmc_pooled_summary$variable
historical_param_names <- mcmc_historical_summary$variable
new_params <- setdiff(pooled_param_names, historical_param_names)
new_params
```
We see that the additional parameters correspond to the baseline value, which is supposed to differ between studies: both the mean `mu` parameter and the standard deviation `omega` parameter gain an additional second dimension here with the addition of the second study.
Let's look at the estimates of these new parameters and compare them between the studies:
```{r}
#| label: summarize_new_params
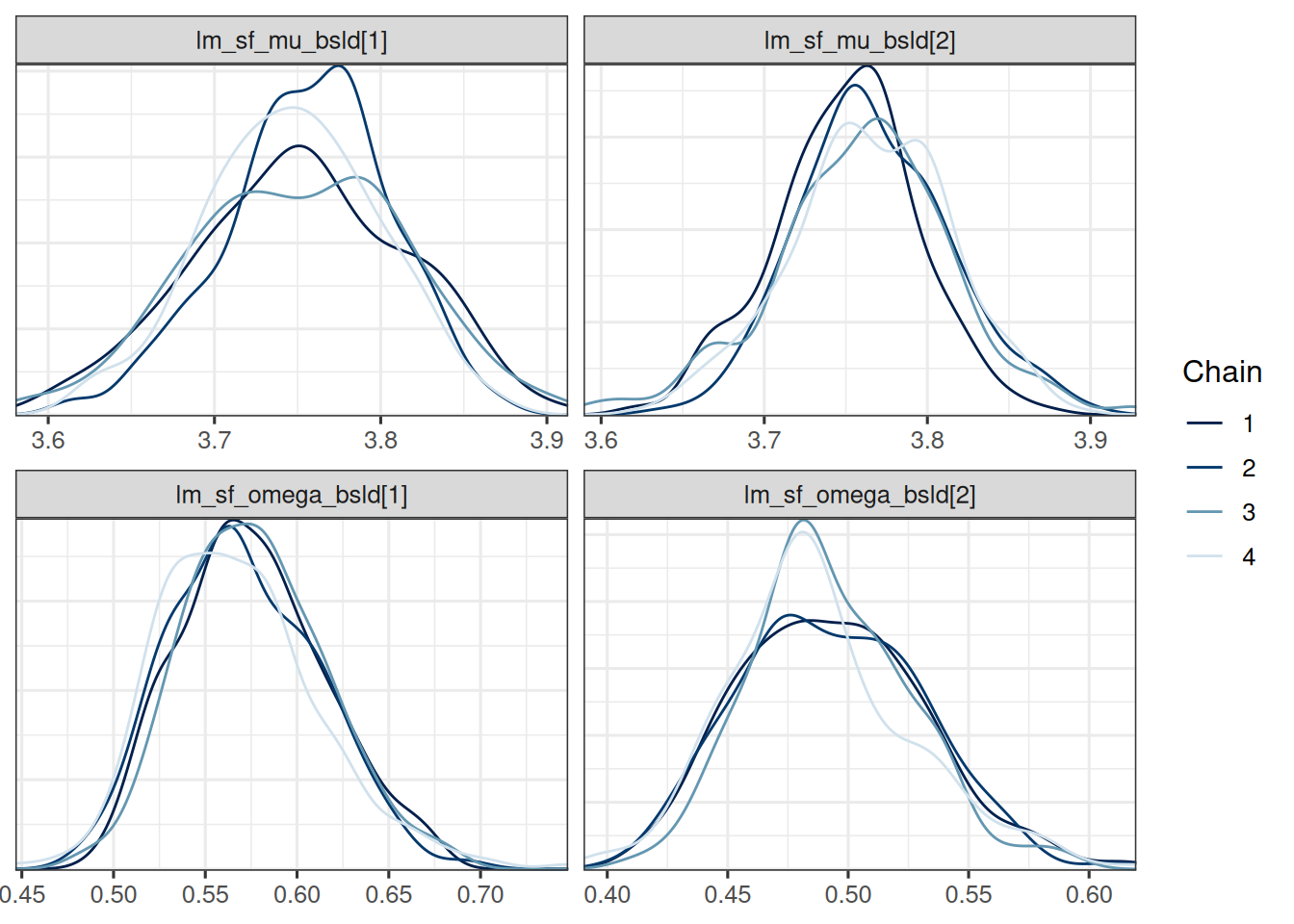
baseline_vars <- c("lm_sf_mu_bsld", "lm_sf_omega_bsld")
baseline_pooled_joint_results <- mcmc_pooled_joint_results$draws(baseline_vars)
mcmc_dens_overlay(baseline_pooled_joint_results)
library(posterior)
# Convert draws to rvars for easier manipulation
baseline_rvars <- as_draws_rvars(baseline_pooled_joint_results)
mu_diff <- baseline_rvars$lm_sf_mu_bsld[1] - baseline_rvars$lm_sf_mu_bsld[2]
summary(mu_diff)
omega_diff <- baseline_rvars$lm_sf_omega_bsld[1] - baseline_rvars$lm_sf_omega_bsld[2]
summary(omega_diff)
```
As expected we don't see a big difference in the baseline SLD between the historical and current study, neither in the mean nor in the standard deviation.
## Goodness of fit
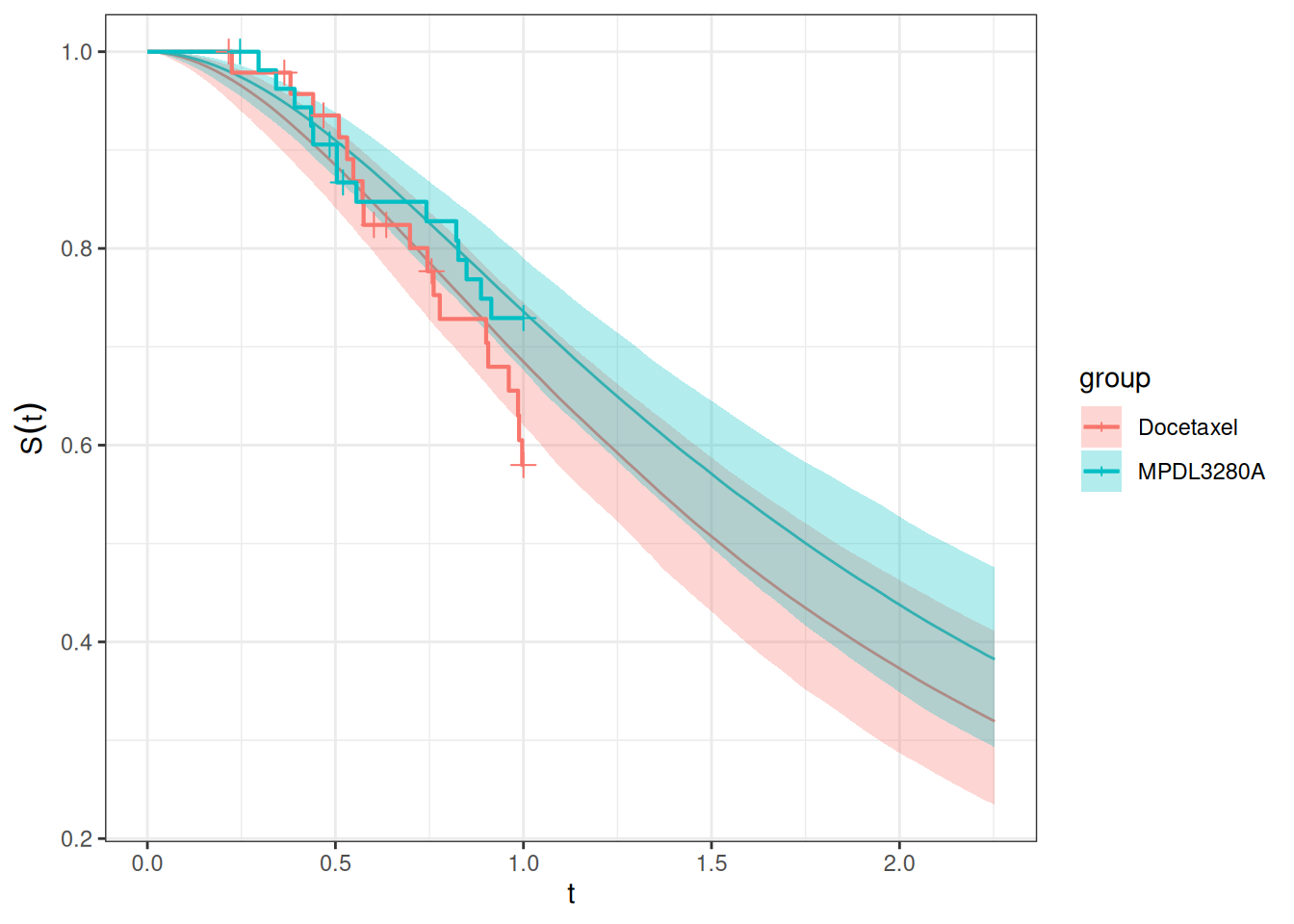
Let's check the goodness of fit to our current study data:
```{r}
#| label: gof_pooled_model
current_subj_df <- current_os_data |>
select(study, id, arm)
time_grid <- seq(from = 0, to = max(pooled_os_data$os_time), length = 100)
current_os_surv_group_grid <- GridGrouped(
times = time_grid,
groups = with(
current_subj_df,
split(as.character(id), arm)
)
)
current_os_surv_pred <- SurvivalQuantities(
object = pooled_joint_results,
grid = current_os_surv_group_grid,
type = "surv"
)
autoplot(current_os_surv_pred, add_km = TRUE, add_wrap = FALSE)
```
So the fit looks satisfactory, considering that the current data are quite immature.
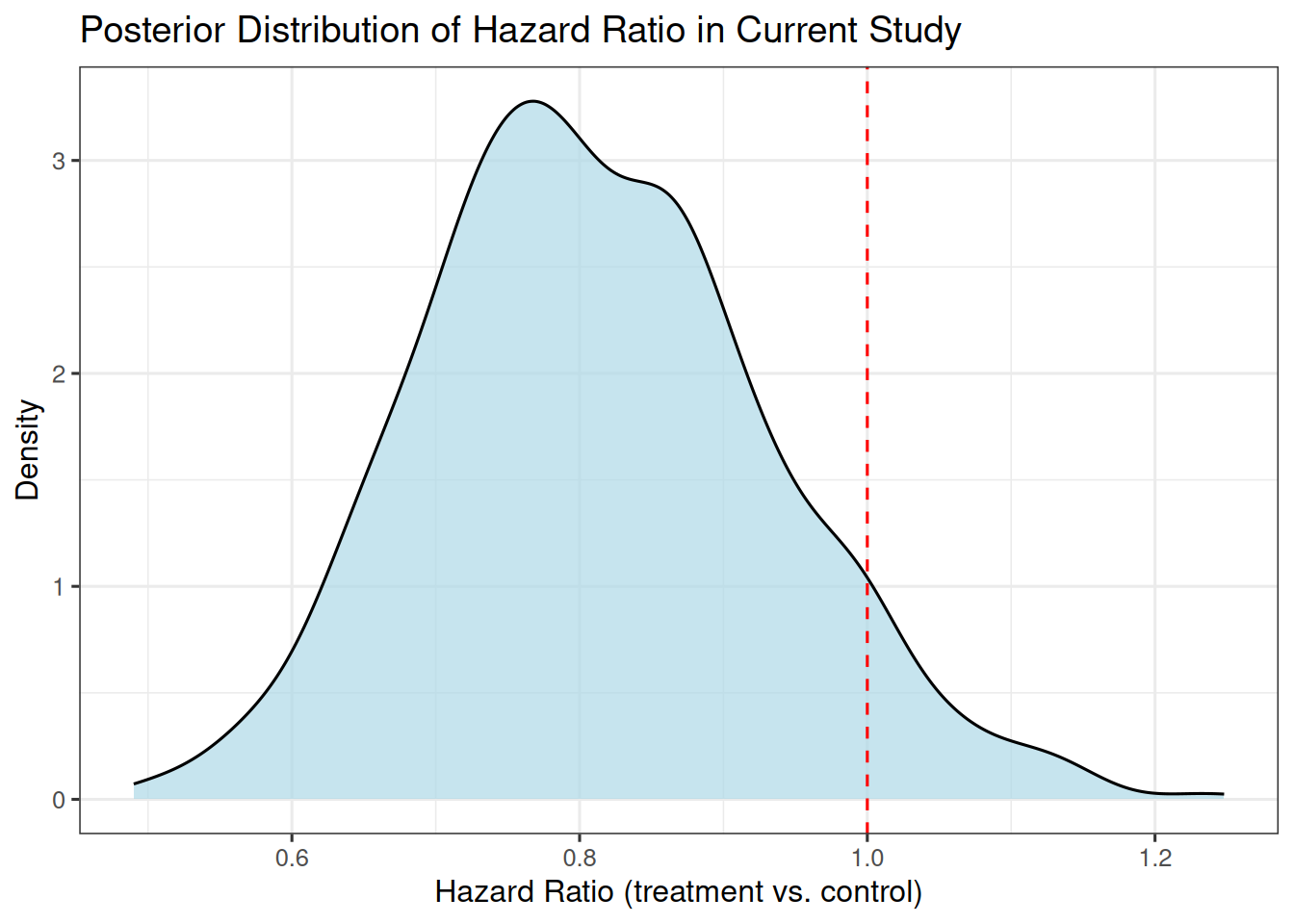
## Posterior distribution of the hazard ratio
Now let's look at the posterior distribution of the hazard ratio between the two treatment arms in the current study.
We do this as follows:
- For a given MCMC sample $i$:
- Sample predicted survival times for the current study subjects which are still being followed up
- Fit a Cox proportional hazards regression model
- Extract the hazard ratio estimate
- Repeat for all MCMC samples to get the posterior distribution of the hazard ratio
This is the same algorithm we used in the session 4 [here](https://rconis.github.io/tgi-os-training/session-pts/1_pts-jmpost.html#individual-samples-based-predictive-power).
In this example we assume for simplicity:
- no additional patients will be enrolled, i.e. we don't need to add new patients to the data set
- all currently censored patients will be followed up until event occurs
Therefore we can directly jump into the sampling step as described [here](https://rconis.github.io/tgi-os-training/session-pts/1_pts-jmpost.html#rcpp-implementation-for-all-patients-and-samples):
```{r}
#| label: sample_current_censored_os_times
# Determine for which patients we want to sample the individual survival times.
pt_to_sample <- pooled_os_data |>
filter(study == "current" & os_event == 0)
pt_to_sample_ids <- pt_to_sample$id
# Obtain the survival function samples (for all patients here).
length_time_grid <- 100
time_grid_end <- round(max(pooled_os_data$os_time) + 10, 1)
time_grid <- seq(0, time_grid_end, length = length_time_grid)
save_file <- here("session-bhm/bhm1_surv_samples.rds")
if (file.exists(save_file)) {
os_surv_samples <- readRDS(save_file)
} else {
os_surv_samples <- SurvivalQuantities(
object = pooled_joint_results,
grid = GridFixed(times = time_grid),
type = "surv"
)
saveRDS(os_surv_samples, file = save_file)
}
qs <- os_surv_samples@quantities
include_qs <- qs@groups %in% pt_to_sample_ids
qs_samples <- qs@quantities[, include_qs]
qs_times <- qs@times[include_qs]
qs_groups <- qs@groups[include_qs]
surv_values_samples <- matrix(
qs_samples,
nrow = ITER * length(pt_to_sample_ids),
ncol = length_time_grid
)
surv_values_pt_ids <- rep(
head(qs_groups, length(pt_to_sample_ids)),
each = ITER
)
censoring_times <- pt_to_sample$os_time[match(
surv_values_pt_ids,
pt_to_sample_ids
)]
library(Rcpp)
# Use Rcpp function for single patient, single sample.
sourceCpp(here("session-pts/conditional_sampling.cpp"))
cond_surv_time_samples <- sample_conditional_survival_times(
time_grid = time_grid,
surv_values = surv_values_samples,
censoring_times = censoring_times
)
os_cond_samples <- matrix(
cond_surv_time_samples$t_results,
nrow = ITER,
ncol = length(pt_to_sample_ids),
dimnames = list(seq_len(ITER), head(qs_groups, length(pt_to_sample_ids)))
)
```
The next step is again to create the complete OS data sets. Here we just use the current data set and replace the censored times with the sampled times above:
```{r}
#| label: create_complete_os_datasets
os_rows_to_replace <- match(
pt_to_sample_ids,
current_os_data$id
)
os_cond_samples_cols <- match(
pt_to_sample_ids,
colnames(os_cond_samples)
)
os_data_samples <- lapply(1:ITER, function(i) {
# Create a copy of the original OS data
os_data_sample <- current_os_data[, c("id", "arm", "os_time", "os_event", "race", "sex", "ecog", "age")]
# Add the sampled OS times for the patients who are still being followed up.
os_data_sample$os_time[os_rows_to_replace] <- os_cond_samples[
i,
os_cond_samples_cols
]
# Set the OS event to TRUE for those patients, because we assume they have an event now.
os_data_sample$os_event[os_rows_to_replace] <- TRUE
os_data_sample
})
```
Now we can fit a Cox proportional hazards regression model to each of the completed data sets and extract the hazard ratio estimates:
```{r}
#| label: fit_cox_models
hr_est_fun <- function(df) {
cox_mod <- coxph(Surv(os_time, os_event) ~ arm,
data = df
)
exp(unname(coef(cox_mod)))
}
```
We can try on one data set first:
```{r}
#| label: test_hr_estimation
hr_est_fun(os_data_samples[[1]])
```
Now we can apply this to all data sets:
```{r}
#| label: estimate_hr_posterior
hr_estimates <- sapply(os_data_samples, hr_est_fun)
```
We can visualize this:
```{r}
#| label: plot_hr_posterior
library(ggplot2)
hr_df <- data.frame(hr = hr_estimates)
ggplot(hr_df, aes(x = hr)) +
geom_density(fill = "lightblue", alpha = 0.7) +
geom_vline(xintercept = 1, linetype = "dashed", color = "red") +
xlab("Hazard Ratio (treatment vs. control)") +
ylab("Density") +
ggtitle("Posterior Distribution of Hazard Ratio in Current Study")
```
## Probability of success
Let's assume the minimal detectable difference, i.e. the hazard ratio that we would consider clinically relevant, is 0.75.
Then our probability of success is given by the posterior probability that the hazard ratio is below 0.75:
```{r}
#| label: calculate_pts
mean(hr_estimates < 0.75)
```